A partir de 2/1/2024, as solicitações devem ser feitas exclusivamente de forma eletrônica, através do Sistema SEI.
A Anvisa informa sobre a alteração no procedimento para solicitar o parcelamento de débitos, a partir de 2/1/2024. A medida tem como objetivos aprimorar e simplificar os serviços prestados.
Atualmente, existem formas distintas de solicitar o parcelamento de débitos junto à Agência. A novidade é que a porta de entrada dessas solicitações será padronizada, com o objetivo de unificar o canal de atendimento e tornar os processos mais simples.
As solicitações devem ser feitas exclusivamente de forma eletrônica, através do sistema SEI, conforme demonstrado nas orientações disponibilizadas em nosso portal a partir desta data.
Eventuais dúvidas poderão ser esclarecidas via Central de Atendimento (0800 642 9782).
Saiba como solicitar o parcelamento de débitos
É importante que o interessado faça previamente a leitura da norma correspondente, a fim de conhecer e cumprir as regras estabelecidas.
- Ao receber a notificação e o boleto emitido pelo Sistema de Cobrança Administrativa (Codiva), o interessado deve acessar o Sistema de Emissão de Boletos e Parcelamento (Sispar), disponível em: <http://www.anvisa.gov.br/SISPAR, utilizando o CNPJ/CPF e o número do débito constante da notificação, para obter a documentação exigida nas normas vigentes, preenchendo-a e assinando-a, além de emitir e pagar a Guia de Recolhimento da União (GRU) constante do Sispar referente à primeira parcela.
- A formalização será feita exclusivamente pelo Sistema Eletrônico de Informações (SEI). Caso o interessado não tenha o cadastro de usuário externo SEI deve providenciá-lo, mediante acesso ao portal da Anvisa – https://www.gov.br/anvisa/pt-br > Sistemas > SEI > Acesso para Usuários Externos (SEI), e seguir as orientações. Para peticionar no SEI, clique em “Peticionamento” > “Intercorrente" e indique o número do processo constante da Notificação. Para informações adicionais, consulte o Manual do Usuário Externo Sei-Anvisa.
- Deve-se juntar ao processo SEI todos os documentos, incluindo a formal solicitação de parcelamento constante do Sispar e/ou da norma correspondente, a qual será analisada pela Gerência de Gestão da Arrecadação.
- Estando em conformidade com a norma, será deferida a solicitação, juntando-se o Parecer ao processo SEI, e serão habilitadas as demais parcelas no Sispar, sendo que estas deverão ser acompanhadas mensalmente e as guias emitidas e pagas diretamente pelo interessado.
- Nos casos em que a documentação não atender a norma, enquadrando-se em uma das hipóteses de indeferimento ou rescisão, o procedimento será realizado mediante o envio da guia para pagamento à vista, ficando vedada a concessão de novo parcelamento.
Para esclarecer dúvidas, entre em contato com a Central de Atendimento da Anvisa, pelo telefone 0800 642 9782, das 7h30 às 19h30, de segunda a sexta, exceto feriados. A ligação é gratuita.
Anvisa aprova mais um produto de terapia avançada para tratamento de câncer no Brasil
O medicamento é mais um produto de terapia gênica que utiliza as células T geneticamente modificadas dos pacientes.
Na reta final do ano de 2023, a Anvisa publicou na seção 1, página 1033, do Diário Oficial da União (DOU) desta sexta-feira (29/12) a aprovação do registro sanitário de mais um produto de terapia gênica à base de células modificadas geneticamente para tratamento do câncer hematológico.
Trata-se do medicamento de terapia gênica TECARTUS® (brexucabtageno autoleucel), do laboratório Kite, uma empresa do grupo Gilead Sciences Farmacêutica do Brasil. Ele é indicado para o tratamento de adultos com linfoma de células do manto (LCM), quando os sintomas ou a doença retornam (recidiva) ou quando não respondem (refratário), após dois ou mais tratamentos anteriores. O LCM é um subtipo agressivo de linfoma não Hodgkin que se desenvolve a partir de linfócitos B anormais. O TECARTUS® é o primeiro produto de terapia avançada indicado para o tratamento desse tipo de câncer raro no país.
O TECARTUS® também foi aprovado no Brasil para o tratamento de leucemia linfoblástica aguda (LLA) recorrente ou sem resposta às terapias anteriores.
Como funciona o TECARTUS®
Este medicamento é mais um produto de terapia gênica que utiliza as células T geneticamente modificadas dos pacientes, de modo a produzirem uma proteína denominada receptor antigênico quimérico (do inglês chimeric antigen receptor – CAR). Essa proteína ajuda as células T a se ligarem a uma proteína presente nas células cancerosas, denominada CD19, para promover a eliminação do câncer do organismo do paciente.
O TECARTUS® é o quarto produto de terapia gênica com células CAR-T aprovado pela Anvisa. Os medicamentos complexos, à base de células CAR-T, pertencem a nova categoria de medicamentos, denominados produtos de terapias avançadas, atuando como imunoterapias personalizadas.
Os dados dos estudos apresentados até o momento demonstraram que pacientes com LCM tratados com TECARTUS® apresentaram uma resposta completa (ou seja, ausência de sinais do câncer), superando os resultados percentuais observados em pacientes após outros tratamentos, no estágio grave da doença. O TECARTUS® também se mostrou eficaz no tratamento da LLA que reapareceu após ou não respondeu a tratamentos anteriores.
Os estudos apresentados destacam que pacientes com LCM, tratados com TECARTUS®, demonstraram uma resposta completa, indicando a ausência de sinais de câncer. Este resultado superou os percentuais observados em pacientes submetidos a outros tratamentos no estágio avançado da doença. Além disso, o TECARTUS® mostrou eficácia no tratamento da LLA que ressurgiu após tratamentos anteriores ou não respondeu a eles.
Cuidados especiais com o paciente
É importante destacar que os resultados de eficácia foram acompanhados de ocorrências de efeitos secundários graves em mais da metade dos doentes, tais como síndrome de liberação de citocinas (SLC, uma doença potencialmente fatal que pode causar febre, vômitos, falta de ar, dor e pressão arterial baixa), encefalopatia (um distúrbio cerebral acompanhado de dor de cabeça, sonolência e confusão mental) e infecções.
As estratégias de monitoramento e mitigação desses eventos adversos são parte fundamental do plano de gerenciamento de risco definido no processo de registro na Anvisa, com medidas de manejo e responsabilização que deverão ser providenciadas para o sucesso da terapia no Brasil.
Entre essas medidas, destacam-se:
- treinamento dos profissionais de saúde envolvidos na manipulação e na administração do produto, no manejo dos eventos adversos relacionados ao uso e na atenção ao paciente;
- qualificação específica para os serviços de saúde que irão coletar e manipular o material de partida (células T do paciente), bem como preparar, administrar e monitorar o paciente;
- educação dedicada ao paciente e familiares com orientações pós-uso do produto;
- rigoroso processo de logística para garantir a manutenção da qualidade e da rastreabilidade de toda a cadeia produtiva do TECARTUS® e de cuidado ao paciente.
Registro na Anvisa
A Anvisa conduziu uma análise criteriosa do TECARTUS®, considerando sua classificação como um medicamento ou produto de terapia gênica, inovador, baseado em células modificadas do tipo CAR-T, para tratamento de doenças raras graves. A avaliação, fundamentada em informações regulatórias e científicas fornecidas pela empresa, abrangeu diversos aspectos, destacando-se:
- Perfil de segurança e prova de conceito: incluiu inúmeros dados de experimentos não clínicos.
- Perfil de segurança e eficácia: utilizou dados de estudos clínicos para analisar a segurança e a eficácia do medicamento em pacientes.
- Processo de obtenção do material de partida: detalhou a metodologia padronizada de coleta e criopreservação das células T do paciente.
- Produção em larga escala: considerou a produção em larga escala, garantindo requisitos de qualidade e boas práticas de fabricação. Destaca-se que o processo produtivo do TECARTUS® é semelhante ao do produto YESCARTA®, também fabricado pela empresa Kite/Gilead.
- Estudos de estabilidade e distribuição: envolveu demonstração de dados da estabilidade do produto no Brasil e da definição dos processos controlados de exportação/importação.
- Estratégias para cuidados ao paciente: incluíram orientações e precauções para cuidados especiais ao paciente.
- Mecanismos de monitoramento e gerenciamento de riscos: abordaram o acompanhamento pós-administração, farmacovigilância e outros requisitos aplicáveis.
A Anvisa certificou os processos de fabricação, incluindo a produção dos vetores virais, com a obtenção da Certificação de Boas Práticas de Fabricação (CBPF) após inspeções nos EUA e na Holanda.
Assim, após avaliações e inúmeras interações com a empresa detentora do registro no Brasil, foi possível demonstrar que o TECARTUS® se apresenta como uma opção de tratamento para LCM e LLA, recidivado e refratário, em situações clínicas graves. Os eventos adversos são controláveis se forem implementadas medidas adequadas de manejo. Portanto, a Anvisa concluiu que os benefícios do produto são superiores aos seus riscos, sendo aprovado seu registro no Brasil.
Foi estabelecido um Termo de Compromisso para o acompanhamento de longo prazo de dados de segurança e eficácia do produto. Isso significa que serão monitorados periodicamente dados adicionais, tanto dos resultados dos pacientes que utilizarem o produto quanto de estudos específicos, que a empresa está obrigada a fornecer à Anvisa. Este tipo de registro condicional é fundamental em produtos de terapia avançada, devido à natureza específica e complexa desse tipo de medicamento. O TECARTUS® também foi aprovado pela agência reguladora dos Estados Unidos (Food and Drug Administration – FDA) e pela European Medicines Agency (EMA), agência reguladora europeia.
CMED divulga os fatores de conversão do Preço Fábrica e do Preço Máximo ao Consumidor referentes às novas alíquotas do ICMS praticadas nos estados
Instrução Normativa publicada visa divulgar os fatores de conversão para a definição do Preço Fábrica e do Preço Máximo ao Consumidor referentes às novas alíquotas de ICMS, sem qualquer alteração de mérito em relação à Resolução vigente.
Foi publicada nesta sexta-feira, 29/12, a Instrução Normativa (IN) 01, de 28 de dezembro de 2023, da Secretaria Executiva da Câmara de Regulação do Mercado de Medicamentos (SCMED). A norma divulga os fatores de conversão do Preço Fábrica (PF) e do Preço Máximo ao Consumidor (PMC) referentes às novas alíquotas do Imposto sobre Operações relativas à Circulação de Mercadorias e sobre Prestação de Serviços de Transporte Interestadual, Intermunicipal e de Comunicação (ICMS) praticadas nos estados de destino.
A IN tem por objetivo divulgar a atualização dos novos fatores de conversão do PF e do PMC previstos nas tabelas constantes dos Anexos I e II da Resolução CM-CMED n. 01, de 30 de março de 2023, em função da introdução de novas alíquotas do ICMS praticadas nos estados de destino. A medida visa orientar a execução da norma pelos agentes públicos envolvidos em seu cumprimento.
Dessa forma, a relação dos fatores de conversão para a definição dos Preços Fábrica e dos Preços Máximos ao Consumidor, previstos nas tabelas constantes dos Anexos I e II da Resolução CM-CMED n. 01/ 2023, fica atualizada com a inclusão das novas alíquotas de ICMS, conforme os Anexos I e II da Instrução Normativa publicada hoje:
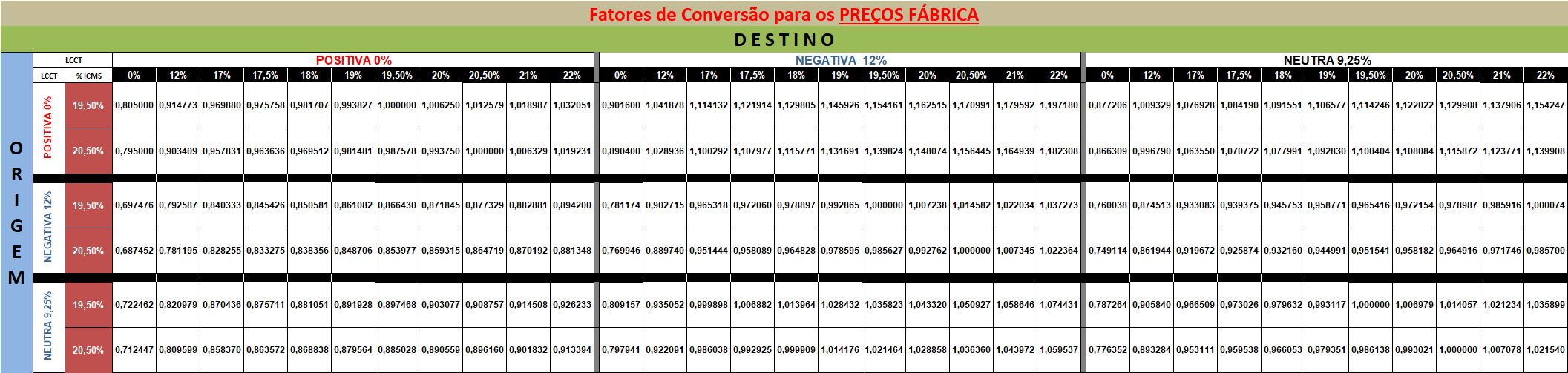
fatores de conversão
Nota explicativa: para a conversão dos preços entre a Lista de Concessão de Crédito Tributário (LCCT) e as diversas alíquotas de ICMS, as empresas deverão utilizar os fatores da matriz acima, partindo sempre do Preço Fábrica (ORIGEM) a ser convertido para o Preço Fábrica (DESTINO), multiplicado pelo fator de conversão correspondente.
I. Preço Origem é o preço a ser convertido.
II. Preço Destino é o preço convertido.
III. Preço Origem X fator de conversão = Preço Destino.

PMC
A SCMED destaca que, até então, a CMED tinha divulgado os fatores de conversão de preços para as alíquotas de ICMS 0%, 12%, 17%, 17,5%, 18%, 19%, 20%, 21% e 22%, conforme pode ser observado nos Anexos da Resolução CM-CMED n. 01/2023.
No entanto, alguns estados da Federação decidiram alterar as alíquotas internas do ICMS, com vigência variável, gerando a necessidade de atualização dos Anexos da mencionada Resolução. Nesse sentido, a IN 01/2023 visa tão somente divulgar os fatores de conversão para a definição dos Preços Fábrica e dos Preços Máximos ao Consumidor referentes às novas alíquotas, sem qualquer alteração de mérito em relação à Resolução do Conselho de Ministros da CMED.
Anvisa cancela novas pomadas para fixar ou modelar cabelos
Produtos foram cancelados por conta de novas regras para pomadas editadas em setembro deste ano.
A Anvisa cancelou 1.266 notificações de pomadas para fixar ou modelar cabelos (Resolução-RE 4.972, de 28/12/2023). A medida faz parte das ações da Agência para garantir produtos seguros, conforme estabelecido na RDC 814, de 1º/9/2023.
A medida tem vigência imediata e esses produtos não podem mais ser comercializados.
Anteriormente, outras resoluções (RE 3.484, de 14/9/2023, e RE 4.290, de 9/11/2023) cancelaram um total de 1.741 pomadas, com base no art. 8º da RDC 814/2023.
Os cancelamentos da resolução publicada nesta sexta-feira (29/12) já estavam planejados como parte das ações contínuas da Anvisa ao longo do tempo e não estão diretamente relacionados aos eventos mais recentes de irritação ocular, que estão em investigação.
Motivo do cancelamento
O artigo 8º da RDC 814/2023 estabelece que serão cancelados processos de regularização de pomadas capilares sem enxágue que atendam certas condições:
- Ter a forma física declarada "pomada".
- Incluir o termo "pomada" no nome ou na rotulagem, em qualquer idioma.
- Ter formulação com 20% ou mais de álcoois etoxilados, incluindo Ceteareth-20 (CAS nº 68439-49-6).
- Ser notificado durante suspensão indicada nos Despachos da Diretoria Colegiada 9 (10/2/2023), 30 (17/3/2023), 31 (22/3/2023) e 59 (19/6/2023).
- As empresas titulares devem ter pelo menos um produto sob sua titularidade temporalmente associado a evento adverso grave notificado à Anvisa.
Registro de produtos de higiene pessoal, cosméticos e perfumes
A partir da RDC 814/2023, as pomadas capilares estão sujeitas a registro na Anvisa, que é o processo em que a Agência precisa avaliar previamente se o produto atende as condições de regulação para estar no mercado. Desde a edição dessa RDC, os produtos notificados no sistema SGAS (Sistema de Automação de Registro de Produtos de Higiene Pessoal, Cosméticos e Perfumes) estão sendo cancelados. A notificação é um processo simplificado de análise e não é mais permitida para essa categoria de produtos.
A fabricação ou comercialização de produtos cancelados e não autorizados constitui infração sanitária sujeita a penalidades, conforme a Lei 6.437/1977.
Quais pomadas estão autorizadas?
A Anvisa destaca que apenas os produtos listados como autorizados podem ser fabricados e vendidos, conforme o artigo 9º da RDC 814/2023. O descumprimento dessa norma é considerado uma infração sanitária, sujeita às penalidades da Lei 6.437/1977. A RE 3.566/2023 proíbe todos os produtos que não estão na Lista de Pomadas Autorizadas
Confira a página com orientações sobre pomadas para trançar ou modelar cabelos.
Problemas com pomadas no Rio de Janeiro
A Agência está trabalhando junto com os órgãos de saúde locais no estado do Rio de Janeiro para entender o problema e sua gravidade. O objetivo é tomar as medidas necessárias para proteger a saúde pública e responder rapidamente aos riscos identificados.
Saiba mais:
- Anvisa alerta consumidores sobre uso de cosméticos nas festas de final de ano
- Anvisa cancela registros de produtos para alisar e ondular os cabelos
- Saiba como consultar um produto de higiene pessoal, perfume ou cosmético
- Anvisa orienta sobre como consultar as pomadas capilares autorizadas
- Usuário ganha plataforma para notificação de eventos adversos
- Anvisa alerta consumidores e profissionais sobre produtos estéticos usados de forma injetável
Fonte: Anvisa, em 29.12.2023.