Condicionantes prévias à importação ainda não foram atendidas pelos estados que firmaram os Termos de Compromisso
Anvisa vem a público atualizar a situação dos processos de importação e noticiar o status de cumprimento das condicionantes para importação relativos aos estados que firmaram os Termos de Compromisso.
Até o momento, foram aprovados os pedidos de importação dos seguintes estados, com os respectivos quantitativos:
Bahia - 300 mil doses;
Maranhão - 141 mil doses;
Sergipe - 46 mil doses;
Ceará - 183 mil doses;
Pernambuco - 192 mil doses;
Piauí - 66 mil doses;
Rio Grande do Norte - 71 mil doses;
Mato Grosso - 71 mil doses;
Rondônia - 36 mil doses;
Pará - 174 mil doses;
Amapá - 17 mil doses;
Paraíba - 81 mil doses;
Goiás - 142 mil doses;
Alagoas - 67 mil doses;
Amazonas - 84 mil doses;
Minas Gerais - 428 mil doses.
Destaca-se que as referidas importações foram autorizadas com exigência de atendimento de 22 condicionantes. Isso se deve às lacunas de informação existentes quanto aos aspectos de qualidade, segurança e eficácia, para permitir uma utilização controlada, segura e inicial da vacina Sputnik V no Brasil.
Quatro estados assinaram os Termos de Compromisso
A deliberação da Anvisa determinou que as condicionantes deveriam constar em Termo de Compromisso a ser celebrado entre a Agência e respectivos governadores e secretários de Saúde, como requisito para o deferimento dos Licenciamentos de Importação (LI) da vacina.
Os Termos de Compromisso foram firmados pela Anvisa, representada pelo diretor-presidente e pelos diretores titulares das diretorias que supervisionam as áreas de Registro de Medicamentos, Fiscalização e Monitoramento.
Até o momento, os seguintes estados providenciaram a assinatura dos termos: Piauí, Bahia, Sergipe e Pernambuco. Os estados do Maranhão, Paraíba, Rio Grande do Norte e Alagoas ainda não assinaram os termos. O estado do Ceará solicitou ajuste para assinatura do termo.
Em relação aos demais estados, a Anvisa não recebeu proposta referente ao cumprimento das condicionantes para elaboração dos respectivos Termos de Compromisso.
Condições
O Termo de Compromisso previu três conjuntos de condicionantes a serem cumpridos pelos importadores:
i) condicionantes prévias à importação;
ii) condicionantes a serem cumpridas durante o processo de importação, licenciamento e internalização da vacina (compreendendo a liberação de lotes pelo Instituto Nacional de Controle de Qualidade em Saúde - INCQS); e
iii) condicionantes que devem ser cumpridas durante o uso da vacina no Brasil.
Até o momento, as três condicionantes prévias à importação não foram atendidas pelos estados que assinaram os termos. São elas:
a) envio de proposição de medida adicional de mitigação do risco pelos fabricantes Generium e UfaVita, decorrente da ausência da validação da etapa de filtração esterilizante;
b) envio do relatório final de validação do processo de fabricação do insumo farmacêutico ativo biológico ou declaração da autoridade russa de que verificou e aprovou tal documento;
c) apresentação, para adequação das Práticas Assépticas e Teste de Esterilidade da UfaVita, dos registros dos treinamentos dos operadores, bem como vídeos demonstrando a execução das atividades críticas nas áreas limpas, indicando que os procedimentos de trabalho foram efetivamente corrigidos e não representam risco de contaminação ao produto.
Critérios técnico-operacionais para análise do INCQS
Sobre o item ii, a Diretoria Colegiada da Anvisa definiu critérios técnicos e operacionais necessários às análises para liberação dos lotes da vacina pelo INCQS, uma vez que o cumprimento da condicionante de nº 6 – Os lotes das vacinas importados somente poderão ser destinados ao uso após análise laboratorial e liberação pelo INCQS – constante na decisão gerou dúvidas de interpretação e também questionamentos de natureza operacional por parte dos importadores e do INCQS.
Dessa forma, foram definidos critérios a fim de tornar exequível a realização dos ensaios de identificação e potência pelo ELISA (teste sorológico imunoenzimático), ensaio de identificação por PCR, ensaio de quantificação de proteína residual e ensaio de segurança específica.
Outro aspecto avaliado pela Agência se refere à possibilidade de que as análises sejam realizadas nas instalações da empresa Inovat Indústria Farmacêutica Ltda., organização empresarial vinculada à União Química, em conformidade com as Resoluções da Diretoria Colegiada (RDCs) 390/2020 e 73/2008, desde que cumpridos os requisitos das boas práticas laboratoriais e respeitada a total autonomia do INCQS para avaliação e liberação dos lotes. Tal decisão também levou em consideração que a empresa já possui boa parte dos documentos, insumos e equipamentos necessários às análises.
A Anvisa reforça que, desde a concessão das autorizações, vem trabalhando de forma colaborativa junto ao Consórcio Nordeste, que representa os estados do Nordeste no processo de importação, a fim de orientar e viabilizar o cumprimento das condicionantes pactuadas, conforme pode ser observado no painel analítico divulgado pela Agência.
Anvisa autoriza novo local de fabricação para vacina da Pfizer
A nova planta fabril está localizada nos Estados Unidos
A Anvisa autorizou, nesta quarta-feira (28/7), um novo local de fabricação para a vacina Comirnaty, da empresa Pfizer. A nova planta fabril é a Hospira Inc., localizada em McPherson, no estado norte-americano de Kansas, e está autorizada como local alternativo de envase, embalagem e testes de controle de qualidade.
Isso significa que a cadeia de produção da vacina fornecida para o Brasil poderá incluir esta nova fábrica, o que pode aumentar a capacidade de fornecimento da empresa para o país.
Todas as vacinas e medicamentos fornecidos para o Brasil precisam ter toda a sua cadeia produtiva autorizada pela Agência. Isso significa que cada nova planta fabril incluída precisa estar de acordo com os requisitos de boas práticas de fabricação.
A vacina Comirnaty está registrada no Brasil desde o dia 23 de fevereiro deste ano.
Esclarecimentos sobre estudos com proxalutamida aprovados pela Anvisa
Anvisa esclarece que até o momento foram autorizados dois estudos clínicos a serem realizados no Brasil com a proxalutamida (GT-0918) para tratamento da Covid-19. Os estudos, patrocinados pela empresa Suzhou Kintor Pharmaceuticals, sediada na China, são:
1. ESTUDO MULTIRREGIONAL DE FASE 3, RANDOMIZADO, DUPLO-CEGO, CONTROLADO POR PLACEBO, PARA AVALIAR A EFICÁCIA E A SEGURANÇA DE GT0918 NO TRATAMENTO DE PACIENTES DO SEXO MASCULINO COM COVID-19 LEVE A MODERADO. VERSÃO 2.0, 21MAR2021. PROTOCOLO GT0918-MT-3001. Autorizado em 10 de junho de 2021.
2. ESTUDO DE FASE 3, RANDOMIZADO, DUPLO-CEGO, CONTROLADO POR PLACEBO, PARA AVALIAR A EFICÁCIA E A SEGURANÇA DA PROXALUTAMIDA (GT0918) EM PARTICIPANTES AMBULATORIAIS DO SEXO MASCULINO COM COVID-19 LEVE A MODERADA. VERSÃO 1.0, 18 DE MARÇO DE 2021. PROTOCOLO GT0918-US-3001. Autorizado em 19 de julho de 2021.
Para aprovação desses estudos, foram apresentados dados de eficácia e segurança para indicações de câncer de próstata e de mama conduzidos em outros países.
Na documentação apresentada à Anvisa, foi citado um estudo conduzido no Brasil para Covid-19 (NCT NCT044464290) por iniciativa de pesquisadores independentes, cujos dados não foram disponibilizados. Também foi comunicado à Agência que estava em andamento um estudo em pacientes graves (NCT04738802), mas nenhum relatório ou resultado foi apresentado.
Esses estudos não foram considerados para a anuência da Anvisa para a pesquisa clínica a ser realizada e também não poderão ser submetidos para fins de eventual registro do medicamento, pois se tratam de estudos cujos dados não são passíveis de avaliação regulatória.
Considerando os dados apresentados e o delineamento dos estudos aprovados para avaliar a segurança e a eficácia da proxalutamida, o perfil de benefício x risco se mantém favorável para a continuidade dos estudos, até que novos dados sejam apresentados.
Anvisa recebe pedido de uso emergencial de medicamento Tofacitinibe
As primeiras 24 horas serão utilizadas para fazer uma triagem do processo.
AAnvisa recebeu nesta quarta-feira (28/7) a solicitação de autorização temporária de uso emergencial do medicamento XELJANZ ®️ (citrato de tofacitinibe). O pedido foi apresentado pela empresa Pfizer Ltda., para que o medicamento já utilizado para o tratamento de artrite reumatóide, artrite psoriática e colite ulcerosa, possa ser autorizado no país para o tratamento da Covid19.
As primeiras 24 horas serão utilizadas para fazer uma triagem do processo e verificar se os documentos necessários para avaliação estão disponíveis. Se houver informações importantes faltando, a Anvisa pode solicitar as informações adicionais ao laboratório.
Análise e prazo
A análise do pedido de uso emergencial é feita por uma equipe multidisciplinar que envolve especialistas das áreas de Registro, Monitoramento e Inspeção de medicamentos. A equipe vem atuando de forma integrada em todos os processos de avaliação de medicamentos e vacinas para combate à Covid-19.
De acordo com a Resolução da Diretoria Colegiada (RDC) 475/2021, que regulamenta o uso emergencial, o prazo de análise do pedido para medicamentos é de 30 dias.
O prazo de avaliação do pedido de uso emergencial não considera o tempo do processo em status de exigência técnica, que é quando o laboratório precisa responder questões técnicas feitas pela Agência dentro do processo.
A Anvisa atua conforme os procedimentos científicos e regulatórios que devem ser seguidos por aqueles que buscam a autorização de vacinas para serem utilizadas na população brasileira. A norma da Agência que regulamenta o processo de autorização para uso emergencial é a RDC 475/2021.
Anvisa divulga resultados de estratégia do segundo trimestre
Relatório de desempenho da estratégia traz dados relativos ao segundo trimestre de 2021.
A Anvisa informa que já está disponível o Relatório de Desempenho da Estratégia da Agência, referente ao segundo trimestre de 2021. A publicação representa mais um instrumento de transparência e traz o monitoramento das metas estratégicas, dos resultados-chave e dos projetos estratégicos, entre outras informações.
É importante esclarecer que esse monitoramento é realizado em quatro janelas trimestrais. Para a 2ª janela de monitoramento, a Agência coletou informações referentes aos resultados alcançados até junho de 2021 das metas e dos projetos estratégicos do Plano Estratégico (PE) 2020-2023 e dos resultados-chave do Plano de Gestão Anual (PGA) 2021. Essa coleta de dados teve participação de todas as unidades da Anvisa e foi feita até o dia 9 de julho.
Com o resultado, foi possível observar que, em média, 36% das ações estratégicas foram alcançadas até o fim do primeiro semestre (percentual médio de metas estratégicas e resultados-chave alcançados e de pacotes de trabalho concluídos dos projetos estratégicos), sendo esse percentual composto por 27% dos resultados-chave do PGA alcançados, 43% das metas estratégicas do PE alcançadas e 39% dos pacotes de trabalho com conclusão prevista para este ano finalizados. O alcance da estratégia corresponde a um valor 8% maior do que o obtido no trimestre anterior.
Em relação à percepção dos gestores quanto ao alcance da estratégia, do conjunto de 94 metas estratégicas e resultados-chave e 11 projetos estratégicos, 62% foram classificados pelos gestores como satisfatórios, 25% como em alerta e 13% como críticos. Além disso, do total, 54 (57%) foram relatados como impactados pela pandemia de Covid-19, número 7% menor que o do trimestre anterior.
No que diz respeito aos projetos estratégicos, houve uma evolução em relação ao último monitoramento, no qual todos os projetos apresentavam pacotes de trabalho em atraso. Até o fim do segundo trimestre de 2021, cinco projetos estavam com execução em dia, conforme o Gráfico 1. Cabe destacar ainda que o projeto estratégico “P3 – Implementação de programa de monitoramento da qualidade de produtos sujeitos à vigilância sanitária baseado em riscos” foi totalmente concluído nesse trimestre.
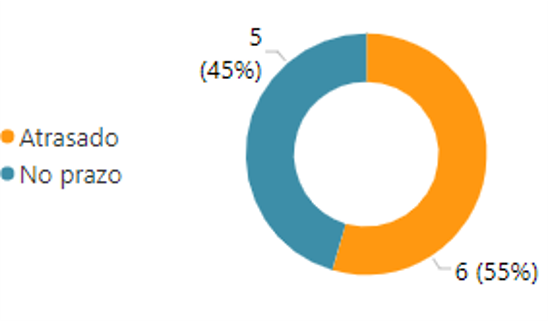
Já em relação às metas estratégicas e aos resultados-chave, 40 (43%) tiveram execução acima de 75% até o segundo trimestre de 2021 (nove metas a mais que no trimestre anterior), conforme o Gráfico 2. Em contrapartida, 36 (38%) apresentaram desempenho inferior a 25% (11 a menos que no trimestre anterior). Não houve mudança na faixa de 25% a 50% em relação ao trimestre anterior. Quanto à faixa de 50% a 75%, há uma a menos nesse trimestre. As alterações significativas apenas nas faixas de execução das extremidades refletem que há um movimento de progresso das metas, saindo da faixa mais baixa e indo diretamente para a de melhor execução.
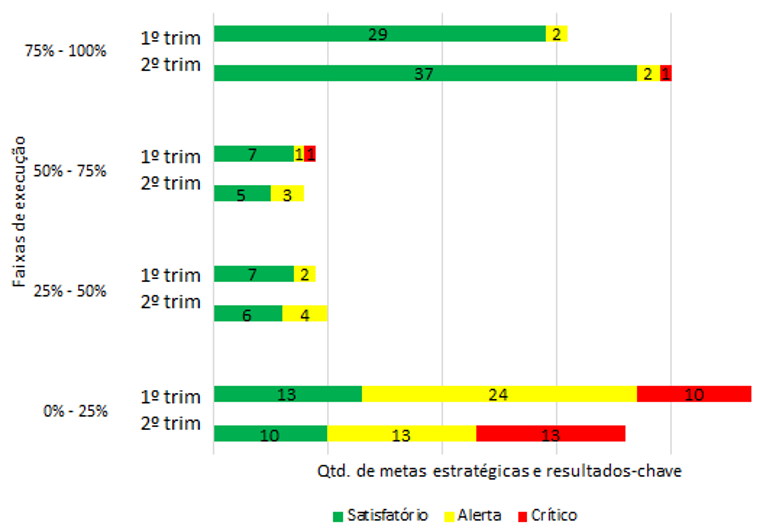
Gráfico 2 – Distribuição das metas estratégicas e resultados-chave conforme faixa de execução, percepção do gestor e trimestre monitorado.
A “categoria de desempenho” (Quadro 1) continuou a ser utilizada nesse trimestre a fim de possibilitar uma visão incremental e sintetizada dessas informações. A combinação da percepção do gestor, do percentual de execução da meta e do trimestre monitorado originam cinco categorias (A, B, C, D e E), de maneira que a melhor (A) indica maior possibilidade de alcance da meta no final do ano e a pior (E) indica menor possibilidade de alcance. A utilização desse indicador é relevante, uma vez que possibilita condensar as informações relevantes para análise da criticidade no que diz respeito ao alcance das metas no fim do período.
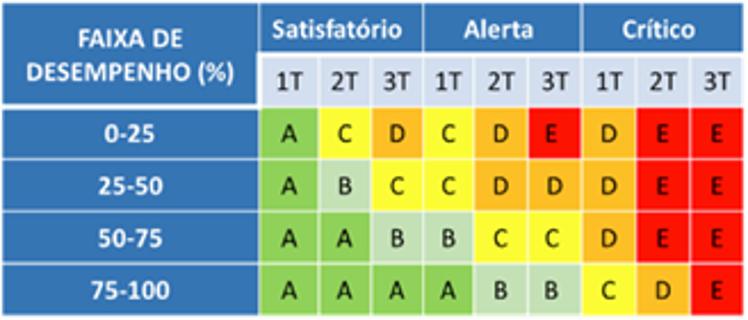
Quadro 1 – Categorias de desempenho.
O Gráfico 3 apresenta a comparação da distribuição das metas estratégicas e dos resultados-chave conforme categoria de desempenho, no 1º e no 2º trimestre de 2021. Segundo o último resultado, 42 (45%) estão na melhor categoria (A), enquanto 13 (14%) estão na categoria E.
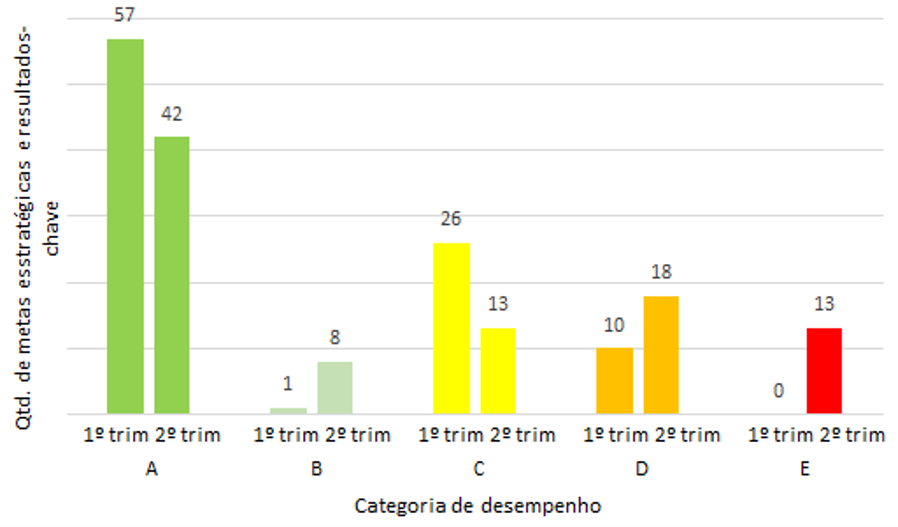
Gráfico 3 – Distribuição das metas estratégicas e resultados-chave conforme categoria de desempenho e trimestre monitorado.
Painel da Estratégia
Lançado em dezembro de 2020, o Painel da Estratégia é uma ferramenta que fornece aos gestores a possibilidade de acompanhamento das metas e dos projetos estratégicos não apenas de suas unidades, mas da Agência como um todo. O painel foi atualizado e já contempla as informações do segundo ciclo de monitoramento. Essa ferramenta tem a finalidade de gerar uma maior internalização da cultura de monitoramento da estratégia e facilitar a tomada de decisão por parte dos gestores.
O Painel da Estratégia possui as visões anual e quadrienal para o monitoramento das metas estratégicas, além de informações atualizadas sobre o monitoramento de todos os projetos estratégicos da Agência obtidas diretamente do PWA (Project Web App, um aplicativo de gerenciamento de projetos utilizado pela Anvisa).
Alerta sobre casos raros de síndrome de Guillain-Barré pós-vacinação
Os eventos adversos foram relacionados às vacinas AstraZeneca, Janssen e CoronaVac. A Anvisa mantém a recomendação pela continuidade da vacinação, uma vez que os benefícios das vacinas superam os riscos. Os fabricantes deverão incluir alerta na bula.
Casos raros de síndrome de Guillain-Barré (SGB) após a vacinação contra Covid- 19 têm sido relatados em diversos países, inclusive no Brasil. Em um comunicado divulgado nesta quarta-feira (28/7), a Anvisa informa que, até o momento, recebeu 27 notificações de casos suspeitos de SGB após a imunização com a vacina da AstraZeneca, além de três casos com a vacina da Janssen e outros quatro com a CoronaVac, totalizando 34 registros.
A SGB é um distúrbio neurológico autoimune raro, no qual o sistema imunológico danifica as células nervosas. Os episódios pós-vacinação (eventos adversos) também são raros, mas já conhecidos e relacionados a outras vacinas, como a da influenza (gripe).
A maioria das pessoas se recupera totalmente do distúrbio. O principal risco provocado pela síndrome é quando ocorre o acometimento dos músculos respiratórios. Nesse último caso, a SGB pode levar à morte, caso não sejam adotadas as medidas adequadas.
É importante destacar que a Anvisa mantém a recomendação pela continuidade da vacinação com todas as vacinas contra Covid-19 aprovadas pela Agência, dentro das indicações descritas em bula, uma vez que, até o momento, os benefícios das vacinas superam os riscos.
Diante dos relatos de eventos adversos raros pós-vacinação, a Agência solicitou que as empresas responsáveis pela regularização das vacinas AstraZeneca, Janssen e CoronaVac incluam nas bulas dos respectivos produtos informações sobre o possível risco de SGB.
Confira a íntegra do Comunicado 008/2021.
Sinais e sintomas
A maior parte dos pacientes percebe inicialmente a SGB pela sensação de dormência ou queimação nas extremidades dos membros inferiores (pés e pernas) e, em seguida, superiores (mãos e braços). Outra característica, percebida em pelo menos 50% dos casos, é a presença de dor neuropática (provocada por lesão no sistema nervoso) lombar ou nas pernas. Fraqueza progressiva é o sinal mais perceptível, ocorrendo geralmente nesta ordem: membros inferiores, braços, tronco, cabeça e pescoço.
Pessoas vacinadas devem procurar atendimento médico imediato se desenvolverem sinais e sintomas sugestivos de SGB, que incluem, ainda, visão dupla ou dificuldade em mover os olhos, dificuldade de engolir, falar ou mastigar. Também devem ficar atentas a problemas de coordenação e instabilidade, dificuldade em caminhar, sensações de formigamento nas mãos e pés, fraqueza nos membros, tórax ou rosto, além de problemas com o controle da bexiga e função intestinal.
Os profissionais de saúde devem ficar atentos para os sinais e sintomas de SGB para garantir o diagnóstico correto, a fim de iniciar os cuidados de suporte e tratamento adequados e descartar outras causas.
Notificação
A ocorrência de SGB pós-vacinação contra Covid-19 deverá ser relatada à Anvisa. É imprescindível o cuidado na identificação do tipo de vacina suspeita de provocar o evento adverso, como número de lote e fabricante.
Profissionais de saúde e cidadãos podem notificar eventos adversos pelo e-SUS Notifica e pelo formulário web do VigiMed. Se o caso for de queixa técnica ou de desvios de qualidade observados em vacinas, seringas, agulhas e outros produtos para saúde utilizados no processo de vacinação, as notificações devem ser feitas pelo Sistema de Notificações em Vigilância Sanitária – Notivisa.
Empresas detentoras de registro ou de autorização temporária de uso emergencial de vacina também devem usar o VigiMed para notificar eventos adversos e o Notivisa para queixas técnicas e desvios de qualidade.
Fonte: Anvisa, em 28.07.2021.