Medida publicada pela Anvisa incentiva registro de radiofármacos no Brasil
Medicamentos são essenciais para diagnóstico e tratamento de doenças como câncer
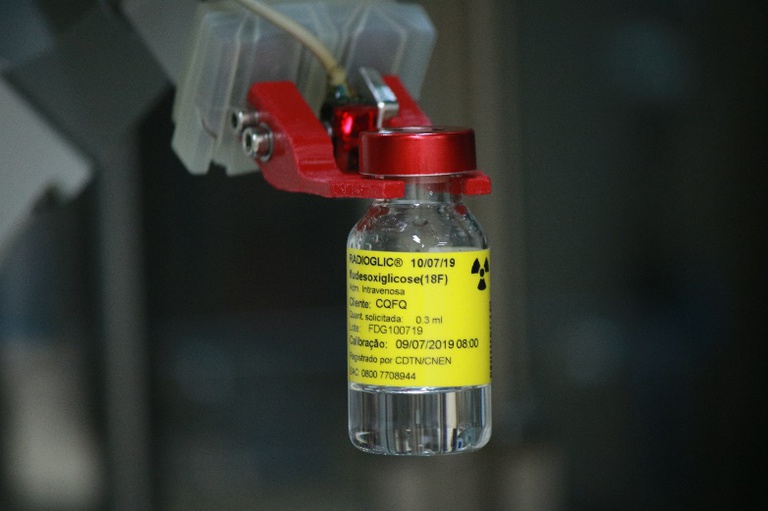
Os radiofármacos são medicamentos utilizados para o diagnóstico e o tratamento de doenças, como o câncer, – Foto: Centro de Desenvolvimento da Tecnologia Nuclear (CDTN)
A Agência Nacional de Vigilância Sanitária (Anvisa) publicou nesta segunda-feira (6/4), a Instrução Normativa (IN) 433/2026, que atualiza a lista de radiofármacos de usos consagrados, com um total de 57 medicamentos. Os produtos previstos nesta relação poderão comprovar sua segurança e eficácia com o uso de dados de literatura científica, nos casos em que tiverem sido estudadas as mesmas atividades e mesmas indicações médicas. A medida garante a ampliação da oferta de radiofármacos à disposição no Brasil.
Relatora do tema, a diretora Daniela Marreco destaca que nos últimos quatro anos o setor de radiofármacos passou a apresentar expansão gradual, com a entrada de novas empresas e produtos. Segundo ela, a atualização dessa lista, que anteriormente previa 33 tipos de radiofármacos, é estratégica, não apenas para refletir a realidade atual do mercado, mas também para apoiar o registro de novos medicamentos e incentivar a inovação.
Os radiofármacos são medicamentos utilizados para o diagnóstico e o tratamento de doenças, como o câncer, que quando prontos para o uso possuem ao menos um radionuclídeo (núcleo) emissor de radiação.
O Guia 61/2023 traz o entendimento técnico da Agência quanto à escolha e apresentação das bibliografias para esses medicamentos.
Atualmente, a regulamentação aplicada a medicamentos radiofármacos é composta, principalmente, por três atos normativos: a RDC 738/2022, que apresenta os requisitos para notificação, registro e importação desses produtos; a IN 319/2024, que detalha a documentação necessária para o registro e a recém-publicada IN 433/2026, que regulamenta a lista de medicamentos passíveis de apresentação de dados de literatura para comprovação da segurança e eficácia.
Histórico
No Brasil a produção de grande parte dos radiofármacos, especialmente os de meia-vida superior a duas horas, foi monopólio estatal até o ano de 2022 e ficava a cargo do Instituto de Pesquisas Energéticas e Nucleares (Ipen), ligado à Comissão Nacional de Energia Nuclear (CNEN).
Durante a pandemia de Covid-19, o país começou a registrar alguns episódios de falta desses medicamentos. Em 2021, para evitar o desabastecimento nacional e manter a continuidade dos tratamentos de saúde dos pacientes brasileiros, a Anvisa autorizou a importação excepcional de radiofármacos sem registro no Brasil, por meio da RDC 567/2021, desde que esses produtos fossem regularizados no país de origem e mediante a responsabilização do importador por critérios de segurança e eficácia. Essa norma perdeu sua validade em 31 de março deste ano.
Ainda, em abril de 2022, com a publicação da Emenda Constitucional 118, o Brasil passou a permitir a produção e a comercialização desses medicamentos por empresas da iniciativa privada.
Anvisa aprova ampliação de uso de medicamento para síndrome hemolítico-urêmica atípica
Nova indicação do Ultomiris® (ravulizumabe) inclui crianças a partir de 5kg
Foto: Freepik
A Agência Nacional de Vigilância Sanitária (Anvisa) aprovou a ampliação de uso do medicamento biológico Ultomiris® (ravulizumabe), usado no tratamento da síndrome hemolítico-urêmica atípica (SHUa). A nova indicação, que teve registro publicado nesta segunda-feira (6/4), estende o uso para pacientes pediátricos com peso corporal a partir de 5kg. Além disso, foi aprovada a remoção da exigência de uso prévio do medicamento eculizumabe antes do início ou da transição para o ravulizumabe.
A SHUa é uma doença rara, grave e potencialmente fatal, caracterizada pela formação de coágulos em pequenos vasos sanguíneos (microangiopatia trombótica), o que pode levar à falência de órgãos, principalmente dos rins. De acordo com o Registro Brasileiro de SHUa, liderado pela Sociedade Brasileira de Nefrologia, entre 2017 e 2020, 75 casos da doença foram registrados no país. A incidência da doença é mais alta em crianças menores de 5 anos e em adultos acima de 65 anos, grupos mais vulneráveis a infecções que podem desencadear a síndrome.
Mecanismo de ação
O medicamento Ultomiris® se liga à proteína C5, bloqueando sua ativação e impedindo a formação do complexo de ataque à membrana – principal mecanismo responsável pelos danos causados na síndrome hemolítico-urêmica atípica (SHUa).
Estudos clínicos confirmaram a exposição sistêmica adequada e a inibição completa do complemento em crianças. Dados demonstraram que o início precoce com ravulizumabe resulta em melhora rápida e sustentada dos desfechos hematológicos e renais, sem a necessidade clínica de terapia anterior com eculizumabe. O perfil de segurança do medicamento foi considerado favorável e consistente com os dados já conhecidos.
Priorização
O pedido de ampliação de uso foi analisado de forma prioritária pela Anvisa, conforme os critérios da Resolução da Diretoria Colegiada (RDC)1.001/2025, por se tratar de uma condição clínica grave e que afeta a população pediátrica.
Leia a Resolução (RE) 1.283/2026 no Diário Oficial da União.
Anvisa manda recolher água oxigenada e proíbe gloss orgânico
Medida foi publicada nesta segunda-feira (6/4) no Diário Oficial da União
Uma ação de fiscalização da Anvisa, publicada nesta segunda-feira (6/3), ordenou o recolhimento da água oxigenada líquida Musa 10 Volumes – lotes que contenham na rotulagem os dizeres: Solução antisséptica, ação antisséptica, para assepsia, fabricados pela empresa Laboratório Musa Ltda.
O produto foi regularizado de forma indevida ao apresentar promessas terapêuticas na rotulagem. Com isso, o item não pode ser comercializado, distribuído, fabricado, divulgado e utilizado.
Gloss orgânico
Outro item atingido pela medida é o gloss orgânico Chuveirinho Liss Therapy Home, de empresa desconhecida. A ação proíbe a comercialização, distribuição, fabricação, propaganda e o uso.
Além de o produto não ter registro sanitário, a empresa não possui autorização de funcionamento.
Confira a Resolução (RE) 1.311/2026, publicada nesta segunda-feira (6/3) no Diário Oficial da União.
Anvisa anuncia novas medidas de combate a irregularidades na importação e manipulação de canetas emagrecedoras
Monitoramento, fiscalização e revisões de normas vão ampliar a proteção e segurança de pacientes que utilizam medicamentos injetáveis de GLP-1

Novas medidas de combate a irregularidades na importação e manipulação de canetas emagrecedoras – Foto: Jacqueline Spotto/Anvisa
A Agência Nacional de Vigilância Sanitária (Anvisa) anunciou, nesta segunda-feira (6/4), novas medidas para garantir a segurança de pacientes que utilizam medicamentos injetáveis de GLP-1 (Peptídeo Semelhante ao Glucagon-1), conhecidos como canetas emagrecedoras, com princípios ativos de semaglutida, tirzepatida e liraglutida.
Entre as medidas previstas estão a revisão das regras atuais do setor, a suspensão das autorizações de funcionamento para farmácias com situação de risco e novas fiscalizações em empresas importadoras de insumos para manipulação.
A Agência anunciou ainda que vai intensificar as ações junto às Vigilâncias Sanitárias dos estados e municípios e realizar acordos de cooperação com agências reguladoras de outros países. Haverá também a criação de um grupo de trabalho com entidades médicas e de saúde.
As medidas ampliam a proteção e a segurança de medicamentos injetáveis de GLP-1 e foram motivadas pelo crescimento irregular da sua manipulação, que pode afetar a saúde dos pacientes. "A questão do controle de qualidade permeia toda a nossa fala, acho que esse é o foco mais importante que é trazer produtos seguros e com garantia de qualidade e eficácia para a população brasileira", destacou o diretor-presidente da Anvisa, Leandro Safatle.
Irregularidades
De acordo com levantamento feito pela Anvisa, a importação de insumos farmacêuticos para a manipulação das canetas tem sido incompatível com o mercado nacional. Somente no segundo semestre de 2025, foram importados mais de 100 kg de insumos, que seriam suficientes para a preparação de aproximadamente 20 milhões de doses.
Outro dado mostra que, em 2026, a Anvisa realizou 11 inspeções em farmácias de manipulação e importadoras, que levaram a 8 interdições por problemas técnicos e falta de controle de qualidade. "A Anvisa tem feito também uma série de fiscalizações de ações nesse sentido. Foram proibidas a importação, o transporte o armazenamento a a comercialização e o uso de produtos irregulares envolvendo a questão dos medicamentos de GLP1", afirmou Safatle.
Entre os riscos mapeados estão a produção sem previsão de demanda por manipulação, problemas de esterilização, deficiências no controle de qualidade e a utilização de insumos farmacêuticos sem identificação de origem e composição.
Desde janeiro deste ano, a Anvisa já publicou 10 ações de proibição de importação, comércio e uso de produtos irregulares que contêm medicamentos agonistas de GLP-1, como semaglutida e tirzepatida.
Para a manipulação de produtos injetáveis, como as canetas, a garantia de padrões rígidos de esterilidade e pureza do insumo é fundamental para garantir a segurança desses produtos para as pessoas.
Pedidos de registro em análise
Atualmente existem oito processos em análise para novos medicamentos com o mesmo princípio ativo do Ozempic, a semaglutida. Desses, sete são de origem sintética e um de origem biológica. Além disso, outros nove produtos aguardam o início da análise pelas áreas técnicas da Anvisa.
A patente da semaglutida no Brasil expirou no dia 20 de março, mas, para que qualquer medicamento possa ser oferecido no mercado nacional, é obrigatória a comprovação de eficácia, segurança e qualidade, que devem ser atestadas pelo registro na Anvisa.
Medidas
São parte do novo plano de ação da Anvisa as revisões das regras atuais para manipulação desses medicamentos, que incluem a Resolução da Diretoria Colegiada (RDC) 67/2007 e a Nota Técnica 200/2025. A Agência também prevê a busca ativa de eventos adversos junto a serviços de emergência, hospitais e clínicas que estejam relacionados ao uso de medicamentos manipulados. Leia o plano de ação.
Confira os eixos de atuação da vigilância sanitária para ampliar a proteção e segurança de pacientes que utilizam agonistas de GLP-1:
Eixo 1 – Aprimoramento regulatório
- Revisão da nota técnica sobre procedimentos para importação, manipulação e controle sanitário de Insumos Farmacêuticos Ativos (IFAs) de agonistas de GLP-1
- Revisão da resolução sobre boas práticas de manipulação de preparações magistrais e oficinais para uso humano em farmácias (RDC 67/2007)
Eixo 2 – Monitoramento e fiscalização
- Intensificação de ações de fiscalização, especialmente de inspeções em importadoras, farmácias de manipulação e clínicas
- Busca ativa de eventos adversos relacionados a medicamentos manipulados, com foco em serviços de emergência, hospitais e clínicas médicas
- Aperfeiçoamento do controle sanitário sobre a importação de IFAs utilizados na produção e manipulação de agonistas do receptor GLP-1
Eixo 3 – Articulação institucional, federativa e internacional
- Criação de grupo de trabalho e celebração de acordo de cooperação com entidades médicas
- Treinamento com o Sistema Nacional de Vigilância Sanitária (SNVS)
- Cooperação com agências reguladoras internacionais
Eixo 4 – Aprimoramento regulatório para análise de petições de registro de agonistas de GLP-1
- Priorização da análise dos cumprimentos de exigências das petições de registro
- Harmonização do uso de guias técnicos de agências reguladoras de referência (ex: EMA e FDA)
Eixo 5 – Comunicação com a sociedade
- Elaboração de plano de comunicação em linguagem simples
- Orientação sobre riscos do uso indiscriminado
- Informação sobre produtos irregulares
- Esclarecimento sobre limites da manipulação magistral
- Campanhas direcionadas a pacientes e profissionais
Eixo 6 – Governança
- Criação de grupo de trabalho na Anvisa para monitoramento e avaliação do Plano de Ação
Reveja a coletiva de imprensa com a apresentação das medidas no canal da Anvisa no YouTube
»Veja mais fotos da coletiva de imprensa no Flickr da Anvisa
Anvisa aprova registro de produto para tratar miastenia gravis generalizada
Rystiggo® é alternativa para pacientes com doença autoimune rara e grave que causa fraqueza muscular
A Agência Nacional de Vigilância Sanitária (Anvisa) publicou, nesta segunda-feira (6/4), o registro de um novo medicamento para tratamento da miastenia gravis generalizada (MGg). Rystiggo® (rozanolixizumabe) é indicado como complemento à terapia padrão de pacientes adultos que apresentam anticorpos específicos (anti-AChR ou anti-MuSK), marcadores da doença que causa fraqueza muscular progressiva e fadiga extrema.
O medicamento age no bloqueio de um receptor responsável pela reciclagem de anticorpos do tipo IgG, mantendo-os em circulação por mais tempo. Ao inibir esse processo, o produto acelera a degradação dos autoanticorpos patogênicos que atacam a comunicação entre nervos e músculos na miastenia gravis. A redução da carga de anticorpos nocivos resulta em uma melhora direta na capacidade funcional dos pacientes.
Os dados que sustentam o registro vêm do estudo clínico de Fase 3 realizado com 200 participantes. Os resultados mostraram melhora funcional e alta taxa de resposta positiva, com taxa acima de 68%. O tratamento se apresentou especialmente eficaz para pacientes positivos para o anticorpo anti-MuSK, grupo que conta com poucas opções de tratamento.
Rystiggo® é uma solução injetável para administração subcutânea, com ciclos de tratamento de seis semanas. O perfil de segurança foi considerado manejável, sendo as reações mais comuns cefaleia, febre e infecções.
Confira a Resolução (RE) 1.283/2026 no Diário Oficial da União.
Anvisa promove 1° Workshop sobre Inspeções em Boas Práticas Clínicas (BPC)
A Agência Nacional de Vigilância Sanitária (Anvisa), por meio da Coordenação de Pesquisa Clínica em Medicamentos e Produtos Biológicos (COPEC) e com o apoio do Sindicato da Indústria de Produtos Farmacêutico (Sindusfarma), realizará, no dia 19 de maio de 2026, o I Workshop de Inspeções de Boas Práticas Clínicas (BPC) da Anvisa. O evento será gratuito e realizado no auditório da Anvisa com transmissão on-line (formato híbrido) e programação voltada a profissionais que atuam na pesquisa clínica no Brasil.
O workshop tem como objetivo promover o diálogo técnico e a disseminação de conhecimentos sobre as inspeções de BPC, com foco especial no cenário atual e na preparação para a implementação dos guias ICH E8(R1) e E6(R3). A iniciativa busca fortalecer a compreensão dos requisitos regulatórios e alinhar expectativas entre os diferentes atores envolvidos no planejamento, desenho e condução de ensaios clínicos.
Ao longo do encontro, serão abordados temas como a renovação do marco das BPC, a experiência da Anvisa na realização de inspeções, tanto de ensaios clínicos com medicamentos (COPEC), quanto de ensaios clínicos com produtos de terapias avançadas – PTAs – (GSTCO), e os principais desafios e oportunidades relacionados à adoção do ICH E6(R3).
O evento contará ainda com a participação de representantes de centros de pesquisa, investigadores, participantes de pesquisa, patrocinadores e organizações representativas de pesquisa clínica (ORPC), além de especialistas na área ética, proporcionando uma visão ampla e integrada sobre o tema.
Entre os objetivos do workshop, destacam-se:
- Ampliar a transparência sobre as inspeções de BPC realizadas pela Anvisa.
- Promover o alinhamento técnico sobre os novos guias internacionais de BPC.
- Estimular o diálogo entre regulador, centros de pesquisa, patrocinadores, participantes de pesquisa e entidades representativas.
- Contribuir para o fortalecimento da segurança do participante de pesquisa e da qualidade e integridade dos ensaios clínicos conduzidos no país.
A programação inclui palestras organizadas por temas e uma sessão final de perguntas e respostas, favorecendo a troca de experiências e o esclarecimento de dúvidas dos participantes.
A Agência reforça que o workshop é uma oportunidade relevante para aprofundar o debate sobre BPC e reafirma seu compromisso com a proteção dos participantes de pesquisa e a qualidade dos dados gerados em ensaios clínicos no Brasil.
Data: 19/5/2026
Local: Auditório da Anvisa (com transmissão on-line) – Endereço: SIA Trecho 05 – Guará, Brasília/DF. CEP 71205-050.
Horário: das 8h30 às 17h
Público-alvo: profissionais que atuam ou tenham interesse no tema de pesquisa clínica
Inscrições para participação presencial: I Workshop de Inspeções de Boas Práticas Clínicas da Anvisa (Presencial Brasília – DF)
Inscrições para participação online: I Workshop de Inspeções de Boas Práticas Clínicas da Anvisa (On-line)
Programação: mais detalhes no link de inscrição
Fonte: Anvisa, em 06.04.2026.